AI-Engine Architecture
How DeepSol Works: The AI Engine
DeepSol is not a simple regression tool; it is an uncertainty-aware ensemble of Deep Neural Networks (DNNs) designed to predict the thermodynamic solubility (LogS) of organic molecules in various solvents at specific temperatures.
Unlike standard models that provide a single "best guess," DeepSol calculates a confidence interval for every prediction, telling you exactly how reliable the result is based on the chemical similarity to its training data.
1. The Architecture: Deep Ensemble with MVE
At its core, DeepSol utilizes a technique called Mean Variance Estimation (MVE).
The Ensemble: Instead of one brain, DeepSol uses 5 independent neural networks trained in parallel. Each network has a slightly different "perspective" on the chemistry.
The Prediction: When you submit a molecule, all 5 networks vote. The final solubility is the average of their predictions.
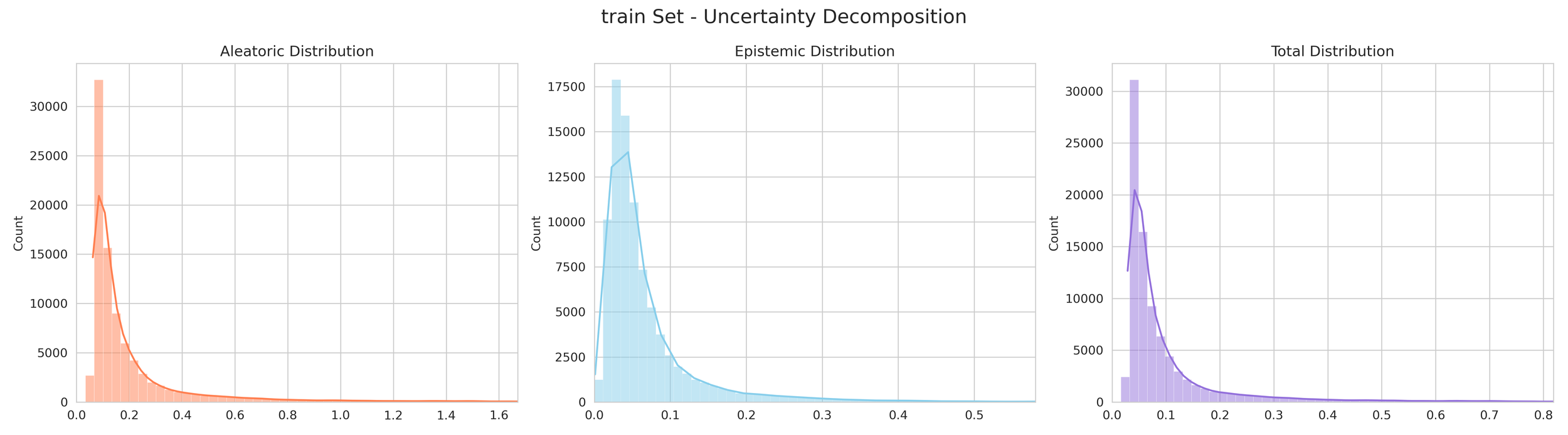
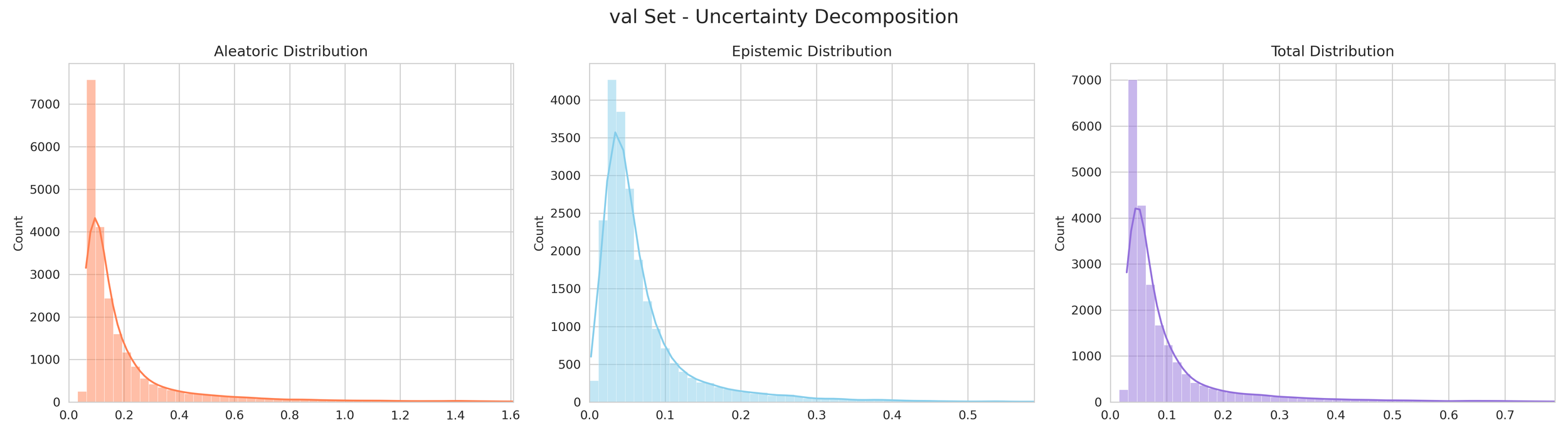
The Uncertainty: The "spread" or disagreement between these networks determines the uncertainty. If all 5 agree closely, confidence is high. If they disagree, the model flags the result as lower confidence.
2. Molecular Representation: 1,200+ Chemical Features
DeepSol "sees" molecules using Mordred Descriptors, a powerful set of mathematical signatures that describe chemical structure.
Input: It takes the Solute SMILES, Solvent SMILES, and Temperature (K).
Processing: It calculates over 1,613 physicochemical descriptors for every molecule—capturing details like atomic connectivity, polarity, ring systems, and electronic topology.
Feature Selection: A specialized "Feature Mask" filters these down to the most critical signals, removing noise and ensuring the model focuses on chemically relevant properties.
3. Training Data & Performance
The model was trained on the AqSolDB and OrgSolDB datasets, covering a vast chemical space ranging from common pharmaceuticals to complex organic compounds.
Target Metric: LogS (Logarithm of Solubility in mol/L).
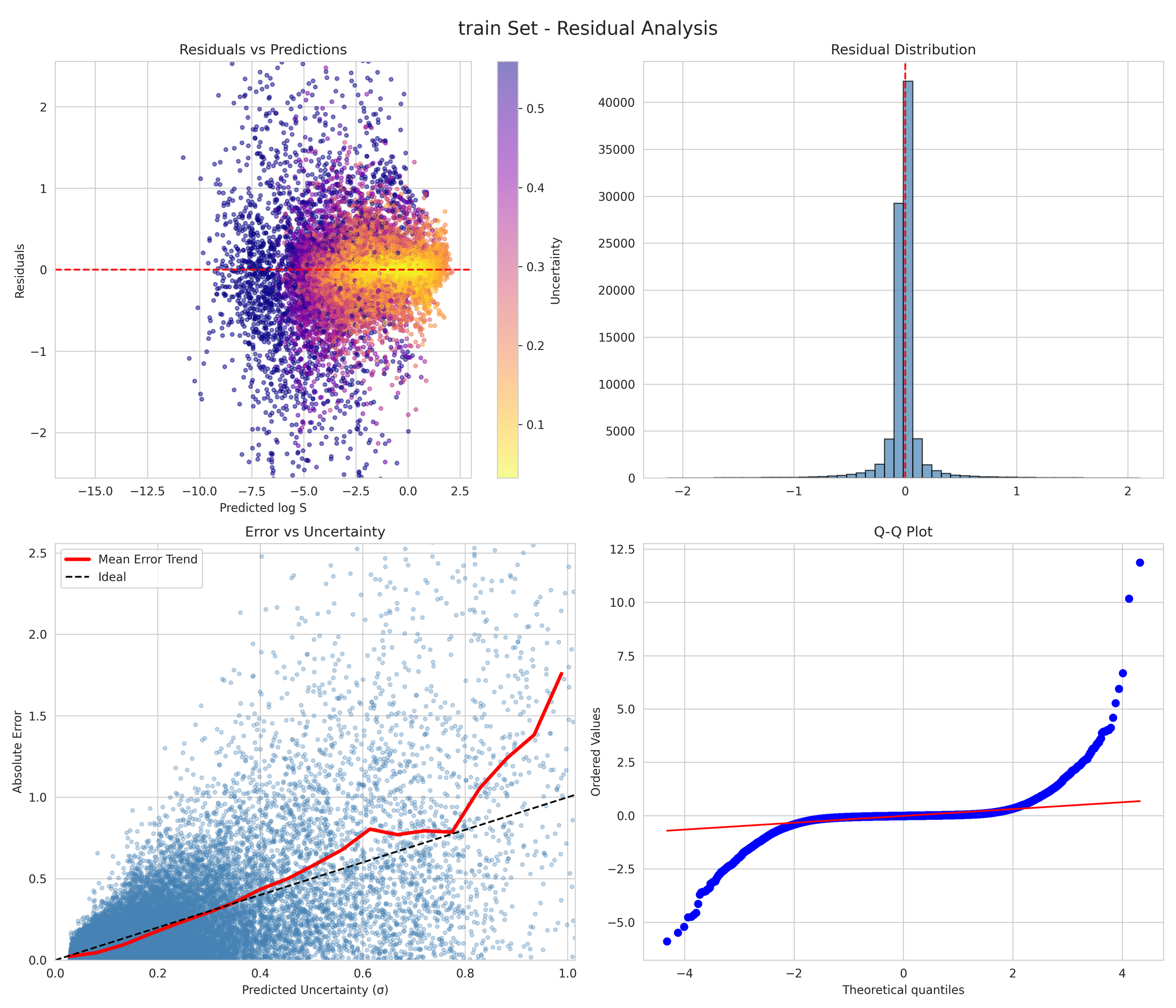
Optimization: The networks were optimized using a Gaussian Negative Log-Likelihood loss function, which forces the model to learn not just the value of solubility, but the variance (uncertainty) inherent in the data.
4. Confidence & Reliability: Beyond Point Predictions
In a laboratory or industrial setting, a single predicted value is only half the story. The real value lies in knowing how much to trust that value. DeepSol’s uncertainty-aware architecture is designed specifically to mitigate the risks of "black-box" AI by providing a transparent reliability score for every result.
The "Confidence-First" Advantage
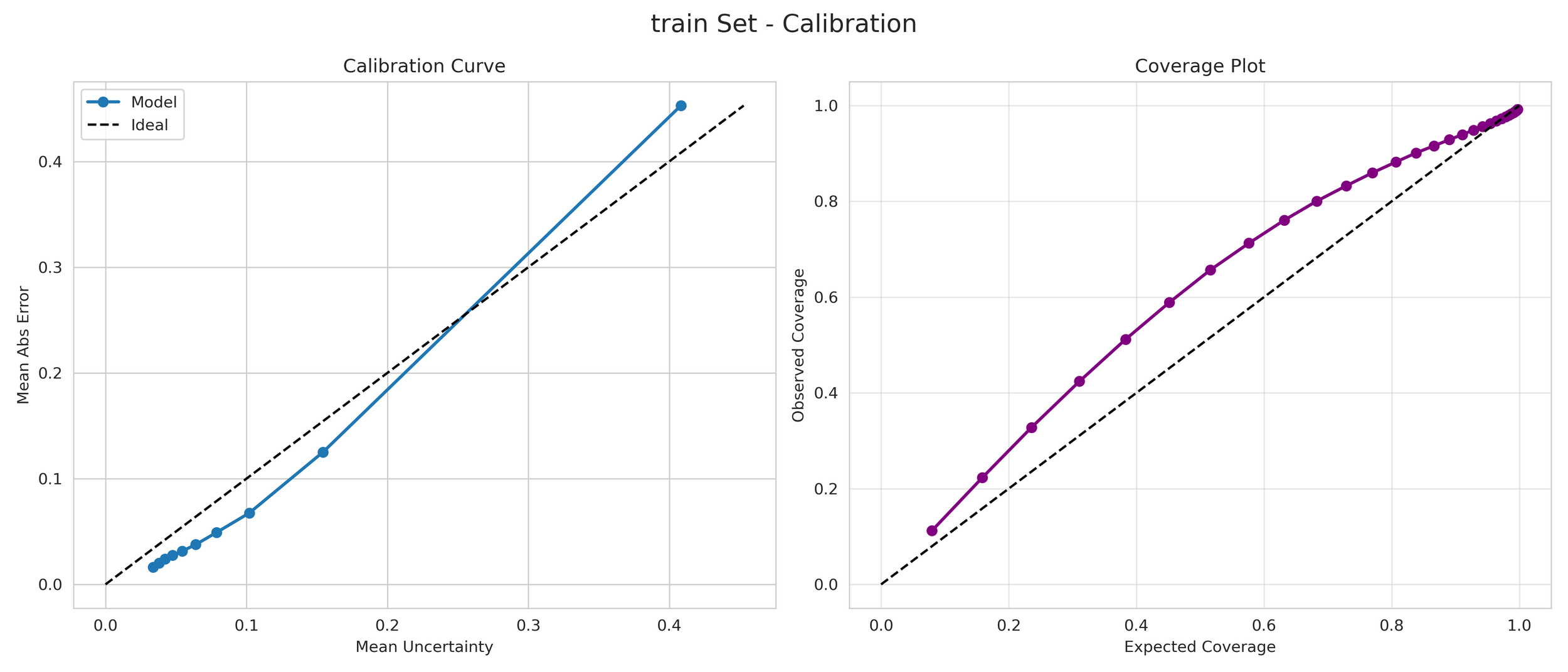
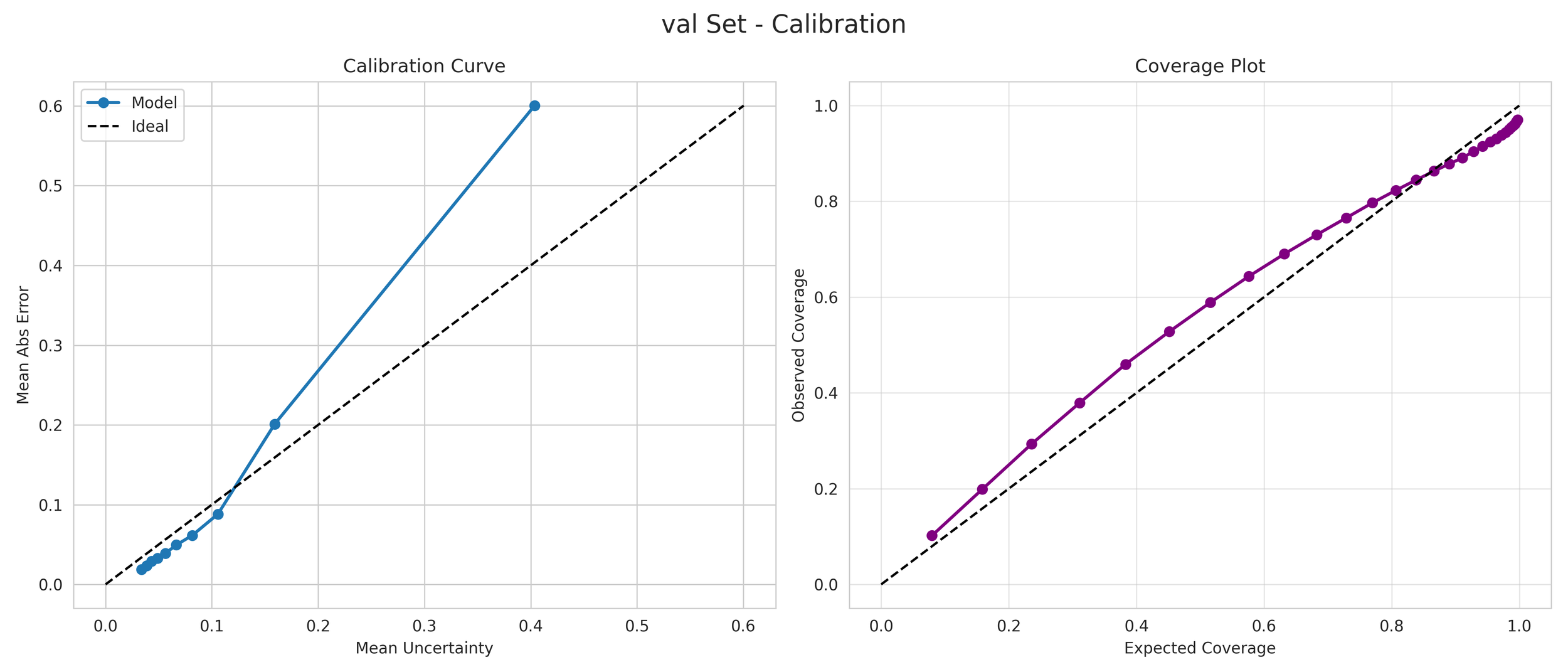
Unlike standard models that provide a single number regardless of how unfamiliar a molecule may be, our Mean Variance Estimation (MVE) framework generates a calibrated 1-sigma prediction interval.
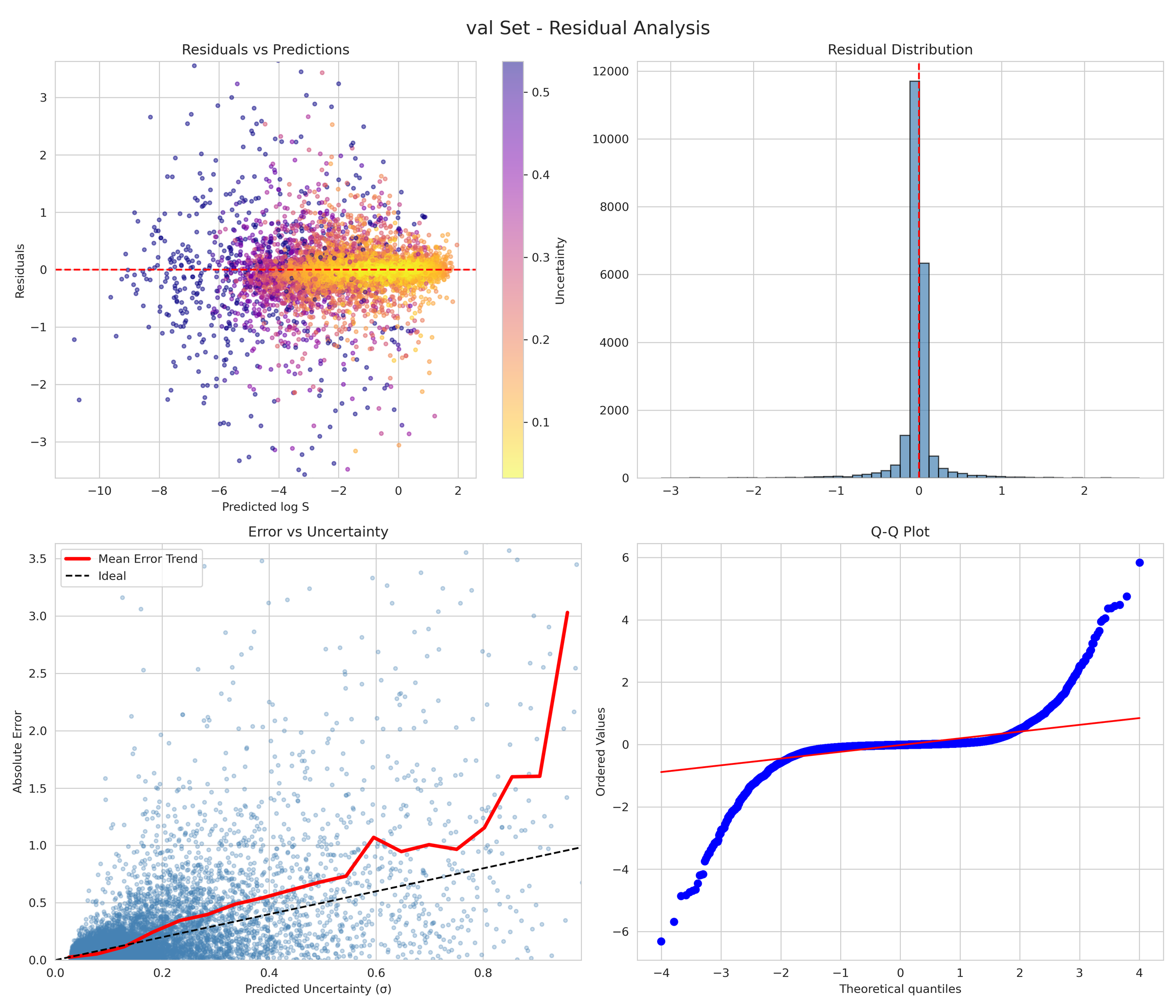
Actionable Uncertainty: If the model predicts a property with high variance, it proactively flags the result as being in a "low-confidence" region of the chemical space.
Risk Mitigation: This allows researchers to prioritize molecules with high-certainty predictions, saving significant time and resources by avoiding experimental validation on unreliable data.
5. Proven Generalization
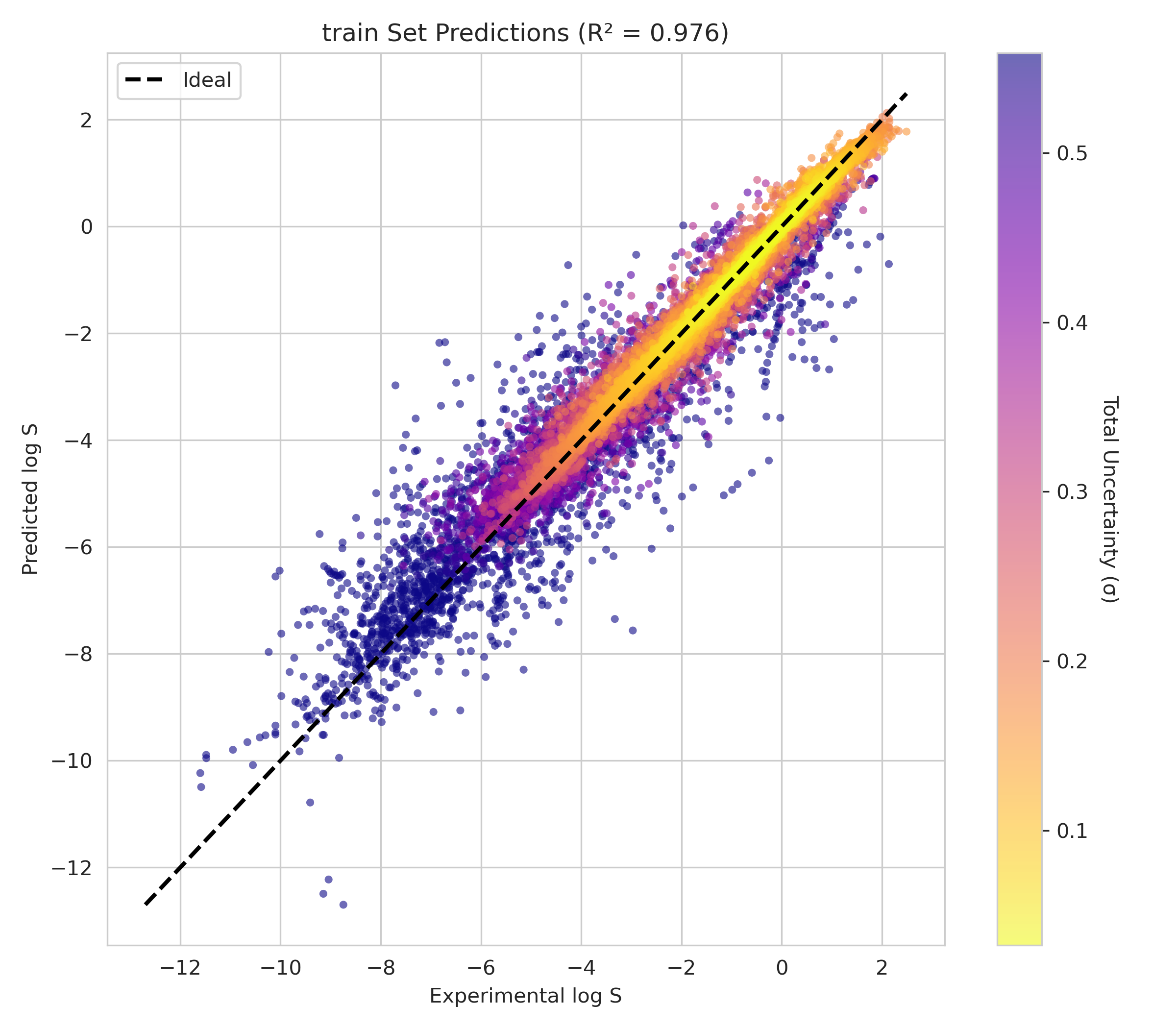
A key indicator of a model's commercial readiness is its ability to perform consistently on data it has never seen. Our current models exhibit a negligible generalization gap—the difference between our training and validation performance is minimal.
Adaptability: This consistency proves that DeepSol has moved beyond pattern matching to internalizing the fundamental physics of molecular interactions.
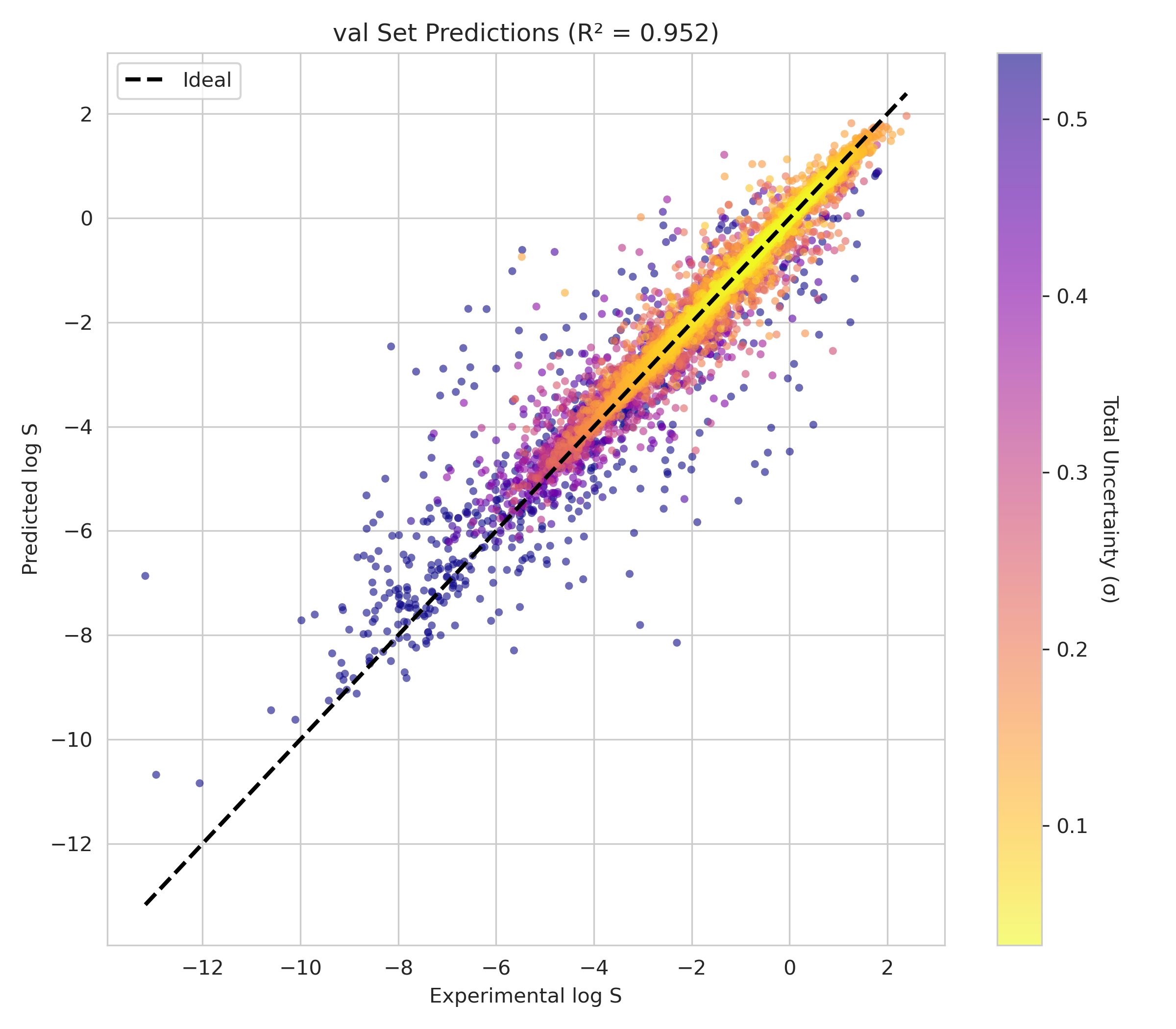
Consistency: Whether you are analyzing a common organic compound or a novel hybrid mixture, the model maintains its high R^2 performance, ensuring that the accuracy you see in our benchmarks is the accuracy you get in your workflow.
Reliability Note: Our 5-model ensemble approach ensures that "outlier" predictions from any single neural network are smoothed out, providing a stable, consensus-based output that outperforms single-model architectures used in standard academic tools.
6. Transparency and Model Metrics
These models demonstrate exceptional robustness and generality by successfully navigating a vast and diverse chemical space, moving beyond simple molecular structures to capture the complex solute interactions of the organic-aqueous/pure and binary domain. The high coefficient of determination values—coupled with the remarkably narrow margin between training and validation performance—serve as a rigorous proof of generalization; they confirm that the models have not merely "memorized" the training data through overfitting, but have instead mastered the underlying chemical principles, making them highly adaptive and reliable for novel, unseen molecular architectures. The graphical performance and uncertainty metrics for the OAP_MPNN-hybrid model are presented below. All available model metrics on www.deepsol.net will be made available upon request, or directly on the website. See the METRICS section for additional model reports and plots.